![]()
![]()
![]()
"SGLT2 inhibitors should also be made available to those with highest risk of problem"
Trial design
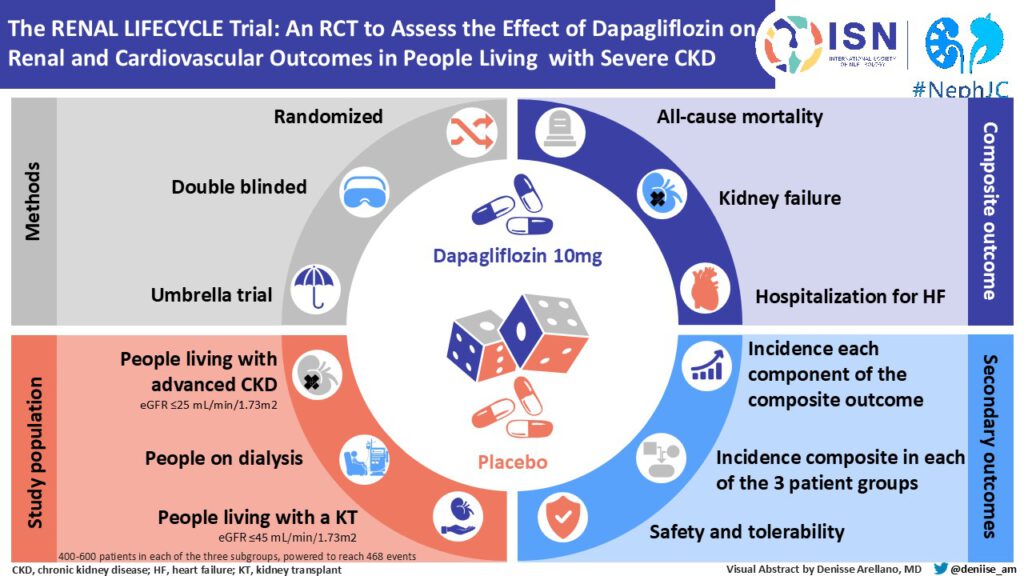
This is a randomized, double-blind, parallel trial. The trial will be conducted multicenter in different geographical regions (the Netherlands, Belgium, Australia and Germany) to make the results widely applicable. Potential participants will be recruited through both academic and non-academic hospitals and dialysis centers. The population chosen for this study is a broad population of patients with severe CKD.
Three strata of patients will be included:
- patients with an eGFR ≤25 mL/min/1.73m2 (non-dialysis or living with a kidney transplant);
- dialysis patients (including hemo and peritoneal dialysis);
- renal transplant recipients with an eGFR ≤45 mL/min/1.73m2.
These patients are almost always excluded from clinical trials even though they are at very high risk of adverse outcomes and few effective therapies are available for these patients.
Patients with type 1 diabetes or those with a life expectancy of less than 6 months according to the treating physician or a planned start of dialysis within 3 months or renal transplantation within 6 months are excluded.
Expectation
We hypothesize that dapagliflozin reduces clinical end points (all-cause mortality, renal failure, and heart failure-related hospitalizations) in patients with stage 4 or 5 CKD, dialysis patients, and renal transplant recipients with and without type 2 diabetes.
Number of participants
1750
Trial duration
18 months recruitment phase, 30 months follow-up after enrollment of last patient: Total study total study duration 48 months. It should be noted that the trial is endpoint-driven and will be terminated when 468 primary composite endpoints have occurred. Therefore, the exact study duration may be shorter or longer than the targeted 48 months.
Inclusion criteria
- Patients with severe renal failure (eGFR ≤25 mL/min/1.73m2) OR
- Hemodialysis and peritoneal dialysis patients (>3 months after dialysis start) OR
- Renal transplant patients with an eGFR ≤45 mL/min/1.73m2 (>3 months after transplantation)
- Age >18 years
- Willing and able to sign informed consent
- Patients with severe renal failure (eGFR ≤25 mL/min/1.73m2) must be on a stable dose of ACE inhibitors or ARBs for at least 4 weeks prior to study start unless there is written evidence that the patient does not tolerate them. Patient maintains dosage during study. This does not apply to the other patient groups.
Exclusion criteria
- Mentally incompetent individuals (ie unable to sign informed consent)
- Type 1 diabetes mellitus
- Current treatment with an SGLT-2 inhibitor
- History with ≥2 urinary tract/genital infections in the past 6 months
- Life expectancy less than 6 months according to attending physician’s assessment
- Planned start of dialysis within 3 months or kidney transplant in the next 6 months
- Patients treated for a renal indication during the last 6 months with a course of systemic immunosuppressive agents or intensification of treatment with systemic immunosuppressive agents, such as patients with a kidney transplant and acute rejection or patients with GPA (Morbus Wegener) and a recent flare
- In renal transplant patients: acute rejection treated with immunosuppressants in the last 6 months
- Active malignancy apart from treated squamous cell or basal cell carcinoma of the skin
- History of severe hypersensitivity or known severe hepatic insufficiency (Child-Pugh class C)
- History of severe non-compliance with medical regimens or unwillingness to comply with the study protocol
- Pregnant or breastfeeding
- Other organ transplants in addition to kidney transplantation
- Severe lactose intolerance
Treatment
Patients take dapagliflozin 10 mg once daily or placebo in the morning according to a randomized treatment regimen. Medication is provided during study visits.
Study design
The trial is pragmatic in design, minimizing the burden on participant and caregiver. Study visits are therefore scheduled in conjunction with regular outpatient visits whenever possible.
Potential participants will be given informed consent, they will be screened by a physician/researcher, and if proven suitable, randomized in a subsequent visit. There is a safety visit after 2 weeks and another visit at 3 months. Thereafter, the frequency of visits will be every 6 months.
Trial related actions will be minimal and largely within regular care. These include for example measurement of vital signs and blood samples.
Summarising